
When you're running a clinical trial, every patient reaction matters-but not every reaction needs to be reported the same way. The difference between a serious adverse event and a non-serious one isn’t about how bad the patient feels. It’s about what actually happened to them. Confusing the two isn’t just a mistake-it’s a system-wide problem that wastes time, delays decisions, and can hide real dangers.
What Makes an Adverse Event 'Serious'?
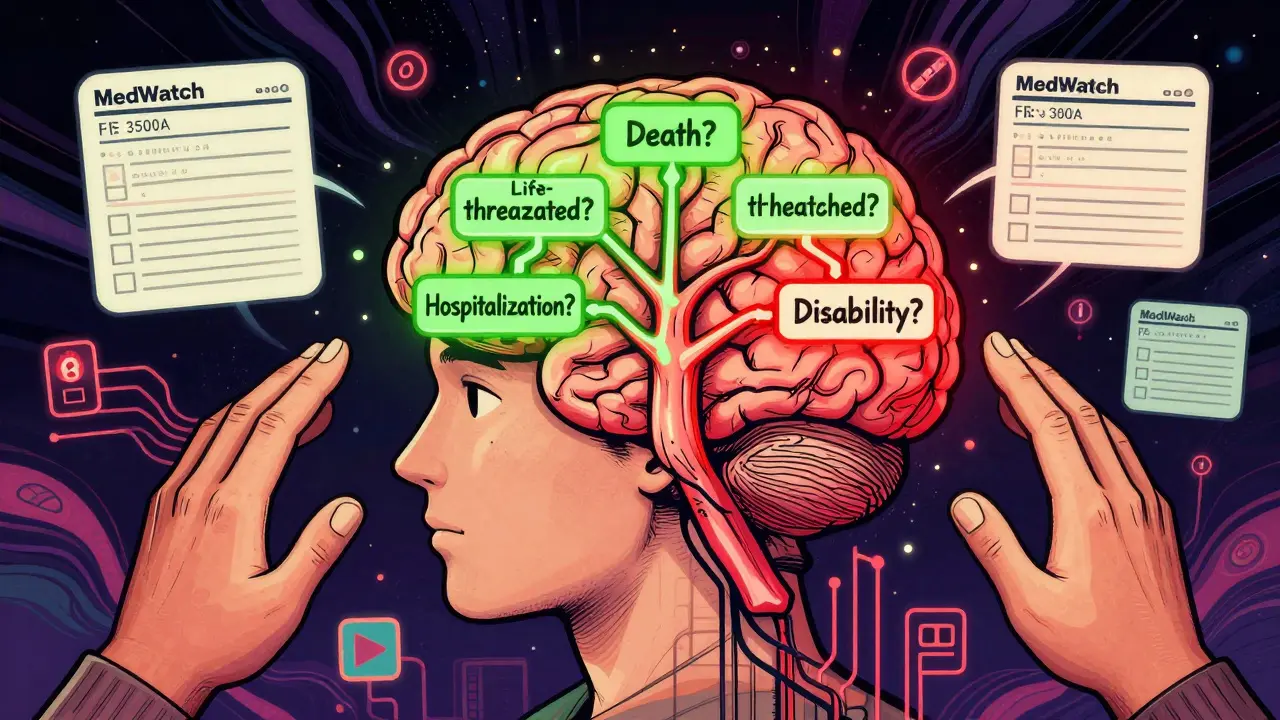
An adverse event (AE) is any unwanted medical occurrence during a clinical trial, whether or not it’s linked to the drug or device being tested. But only some AEs are serious. The FDA and ICH E2A guidelines define seriousness by six specific outcomes, not by how intense the symptoms feel.
A serious adverse event (SAE) is one that:
- Results in death
- Is life-threatening
- Requires hospitalization or extends an existing hospital stay
- Causes persistent or significant disability or incapacity
- Leads to a congenital anomaly or birth defect
- Requires medical or surgical intervention to prevent one of the above
That’s it. No more, no less.
Here’s the part people get wrong: a 'severe' headache is not automatically a 'serious' adverse event. Severe describes intensity-mild, moderate, or severe. Serious describes outcome. A patient could have a severe headache that’s just a bad migraine, resolves in hours, and needs no treatment. That’s not serious. But if that same headache leads to a stroke, hospitalization, or permanent vision loss? That’s serious-even if the initial pain was rated as 'moderate'.
Why the Confusion Happens
It’s not just new staff who mix this up. A 2022 survey of 347 research sites found that 63.4% had inconsistent seriousness determinations across different studies at the same institution. In oncology trials, where patients often enter with pre-existing conditions, the confusion is worse-78.2% of sites reported inconsistency.
Why? Because 'severe' sounds like 'serious.' And in everyday language, they’re used interchangeably. A patient says, 'I had a severe reaction,' and the coordinator hears 'serious.' But 'severe anxiety' during a trial? Unless it leads to self-harm, hospitalization, or inability to function for weeks, it’s not a serious event. Yet in Reddit threads from clinical research coordinators, 89% said they’ve misclassified psychiatric events because of this exact confusion.
Dr. Robert Temple, former FDA deputy director, called this one of the most persistent errors in safety reporting. And it’s costly. In 2022, $1.89 billion was spent across the pharmaceutical industry on adverse event reporting-and 62.7% of that went to processing events that weren’t actually serious. That’s billions spent chasing noise.
When and How to Report
Reporting timelines are strict-and different for each type.
For serious adverse events:
- Investigators must report to the sponsor within 24 hours of learning about the event-even if they think it’s unrelated to the study drug.
- Sponsors must report to the FDA within 7 days for life-threatening events, and 15 days for all other serious events.
- IRBs must be notified within 7 days of the event being identified.
These deadlines are non-negotiable. Miss one, and you risk regulatory action, study suspension, or even legal consequences.
For non-serious adverse events:
- These are documented in Case Report Forms (CRFs) but aren’t reported urgently.
- Reporting happens according to the study’s Data and Safety Monitoring Plan (DSMP)-usually monthly or quarterly.
- Some protocols don’t require IRB reporting of non-serious events at all unless they’re frequent or unusual.
That’s the key: non-serious doesn’t mean unimportant. It means it doesn’t meet the legal threshold for urgent action. You still record it. You still track trends. But you don’t send out an emergency alert.

The Decision Tree That Saves Time
The NIH’s 2018 guidelines offer a simple four-question flow to determine seriousness:
- Did the event cause death?
- Was it life-threatening?
- Did it require hospitalization or extend an existing stay?
- Did it cause persistent or significant disability or congenital anomaly?
If the answer to any of these is yes-it’s serious. Report it immediately. If the answer is no to all four, it’s non-serious. Document it. Don’t report it urgently.
That’s it. No guesswork. No opinions. Just facts.
Many sites now use the FDA’s MedWatch Form 3500A, which has checkboxes for each of these six criteria. It forces you to pick one-no room for ambiguity.
What’s Changing in 2025?
The system isn’t perfect. In 2020, nearly 29% of expedited safety reports submitted to the European Medicines Agency didn’t meet seriousness criteria. In 2022, UCSF’s IRB spent an average of 9.7 business days clarifying each misclassified report.
That’s why change is coming.
The FDA’s May 2023 draft guidance proposes tiered reporting timelines within serious events-so a hospitalization for a known side effect might get a 15-day window, while a sudden cardiac arrest gets 7 days. The European Union’s Clinical Trials Regulation, fully in force since 2022, has already harmonized seriousness definitions across all 27 member states, cutting cross-border reporting errors by 34.8%.
And now, AI is stepping in. Automated tools that analyze AE reports for seriousness are now correctly classifying events in 89.7% of cases-better than human reviewers. MIT’s 2023 study showed these systems could cut processing time by nearly half. But here’s the catch: AI still needs a human to sign off. You can’t outsource judgment. You can only support it.
Training Is Not Optional
ICH E6(R2) requires that every investigator and study coordinator receive documented training on seriousness criteria before a trial starts. And 98.7% of top 50 research institutions require annual refreshers.
Why? Because if one person misclassifies an event, it ripples out. A sponsor gets flooded with false alarms. An IRB wastes time reviewing non-critical reports. A regulator misses a real signal because the system is buried in noise.
Training isn’t a checkbox. It’s a safety net. Use the Common Terminology Criteria for Adverse Events (CTCAE) to grade severity (mild, moderate, severe). Use the six ICH criteria to judge seriousness. Keep them separate. Teach your team to do the same.
What Happens If You Get It Wrong?
Under-reporting a serious event? That’s dangerous. You could miss a pattern that indicates a real risk-like a drug causing unexpected liver failure. The FDA can shut down a trial. A sponsor can lose approval. Lives could be at stake.
Over-reporting? That’s also dangerous. When every minor rash or headache is flagged as serious, the system becomes numb. Regulatory staff stop paying attention. Critical signals get lost. The SWOG Cancer Research Network found that 31.8% of their SAE reports had to be corrected because of wrong seriousness classification-wasting 18.5 full-time hours every week.
There’s no middle ground. You have to get it right.
Final Rule: When in Doubt, Report
There’s one last thing: if you’re unsure whether an event is serious, report it. Better to send a report that turns out to be non-serious than to miss one that is.
But don’t stop there. After you report it, go back. Ask: why was I unsure? Was it the wording? The lack of training? The unclear protocol? Fix the system so the next person doesn’t have to guess.
Safety monitoring isn’t about fear. It’s about clarity. The goal isn’t to report everything. It’s to report the right things-fast. So the right people can act. So patients stay safe. So science moves forward without being drowned in noise.
Is a severe headache always a serious adverse event?
No. A severe headache is only a serious adverse event if it leads to death, hospitalization, life-threatening complications, or permanent disability. If it resolves on its own or with over-the-counter medication and doesn’t disrupt normal function long-term, it’s non-serious-even if it was intense.
Do I report an adverse event if I think it’s unrelated to the study drug?
Yes. You report serious adverse events regardless of whether you believe they’re caused by the investigational product. Causality is determined later by the sponsor and regulators. Your job is to report the event, not judge its origin.
Can an event be serious without hospitalization?
Yes. If an event is life-threatening, causes permanent disability, or requires intervention to prevent death or disability, it’s serious-even if the patient never went to the hospital. For example, a severe allergic reaction treated successfully in the ER with epinephrine and steroids may still qualify as serious if it was life-threatening.
How do I know if an event is 'life-threatening'?
A life-threatening event is one in which the patient was at substantial risk of dying at the time it occurred. It’s not about what might happen later. It’s about the immediate danger. If a patient had cardiac arrest during the trial, that’s life-threatening-even if they were revived. If they had severe chest pain and were admitted for observation but never in danger of dying, it’s not.
Do I need to report non-serious adverse events to the IRB?
Usually not. Most IRBs only require reporting of serious events unless the protocol says otherwise. Non-serious events are tracked internally and summarized in periodic safety reports. Always check your study’s specific reporting plan.
What if my institution doesn’t have clear guidelines?
Follow the FDA and ICH definitions. Use the four-question decision tree: death? life-threatening? hospitalization? disability? If any answer is yes, report it as serious. Document your reasoning. And push your institution to create standardized training-it’s not just good practice, it’s required by global standards.
Are there tools to help classify adverse events correctly?
Yes. Many sponsors now use AI-powered safety platforms that analyze AE narratives and suggest seriousness classification based on ICH criteria. These tools are about 90% accurate, but they still require human review. Use them as assistants, not replacements.
Wendy Lamb
February 4, 2026 AT 12:35Finally, someone laid this out clearly. Too many sites treat 'severe' and 'serious' like synonyms. I've seen coordinators panic over a bad headache-like it's a red alert. Nope. It's a migraine. Document it. Move on.
Prajwal Manjunath Shanthappa
February 5, 2026 AT 14:47Oh, for heaven’s sake-this is why clinical research is a circus! You’ve got people conflating subjective intensity with regulatory thresholds like it’s a poetry slam! The ICH E2A criteria aren’t suggestions-they’re the law! And yet, we still get ‘severe anxiety’ flagged as an SAE because someone read a novel where the protagonist had a nervous breakdown?!
Let me be perfectly clear: if a patient says, ‘I felt really scared during the infusion,’ and then goes home to binge-watch Netflix, that is NOT serious! It’s a feeling! Not a medical outcome! The FDA doesn’t care about your emotional weather report!
And don’t even get me started on the AI tools-they’re brilliant, yes, but they still need a human to sign off? Who signs off on the sign-off? A committee? A mid-level manager who last read a protocol in 2018?!
It’s a house of cards. And we’re all pretending the foundation isn’t made of wet paper towels. I’ve seen sites spend 40% of their budget on false positives. Billions. On noise. While real signals-like that one patient who got liver necrosis after dose 3-get buried under a pile of ‘moderate nausea’ reports.
Training isn’t optional. It’s the only thing standing between a patient’s life and a spreadsheet full of nonsense.
And yes-I’m angry. Because I’ve had to clean up this mess. Again.
Antwonette Robinson
February 7, 2026 AT 08:09So let me get this straight-you’re telling me a 72-year-old with metastatic cancer who gets a 10/10 headache after chemo doesn’t get an SAE unless they end up in the ICU? That’s the rule? Wow. Just… wow. So the system rewards patients who don’t complain. Brilliant.
Ed Mackey
February 7, 2026 AT 23:39Biggest thing i learned? dont assume causality. i once thought a rash was from the drug-turns out it was from the new laundry detergent. i still reported it. turned out fine. but i almost didn’t because i assumed. big mistake.
Alex LaVey
February 9, 2026 AT 20:16For anyone new to this-just remember: it’s not about how the patient feels. It’s about what actually happened to their body. That’s the core. I’ve trained over 200 coordinators in the last five years, and every single one of them had the same confusion at first. It’s not their fault. The words are too similar. But once you internalize the six criteria? It clicks. Like learning a new language. You stop translating ‘severe’ as ‘serious.’ You start thinking in outcomes. And that’s when you become a good safety officer.
Also-use the NIH decision tree. Print it. Tape it to your monitor. Use it every time. Even if you’re tired. Even if it’s 2 a.m. and you’re on call. Use the damn tree.
And yes-AI helps. But it’s not a magic wand. It’s a compass. You still have to know where you’re going.
Joseph Cooksey
February 10, 2026 AT 14:46Oh, sweet merciful Jesus on a pogo stick, here we go again with the ‘severe vs serious’ debacle. It’s like watching a toddler try to assemble IKEA furniture while yelling, ‘I don’t need the manual!’
Let me paint you a picture: a woman in Phase 3 gets a ‘severe’ headache after taking the trial drug. She takes ibuprofen. It goes away. She sleeps. Next day? Fine. But the coordinator, fresh out of undergrad, panics-‘OH MY GOD, SHE HAD A SEVERE HEADACHE-WE MUST REPORT!’
Meanwhile, the real red flag? The guy who had a mild dizziness, didn’t report it, and then had a transient ischemic attack two days later. That’s the one that gets buried under 47 reports of ‘moderate fatigue’ and ‘slight nausea.’
The system is broken because we treat safety like a popularity contest-everyone wants to be the hero who caught the big one, so they report everything, and then we drown in noise. The FDA isn’t looking for drama. They’re looking for signals. And signals don’t scream-they whisper. And we’re too busy shouting to hear them.
And don’t even get me started on the IRBs. They spend 9.7 days per misclassified report? That’s 9.7 days of human life wasted on paperwork instead of saving lives. We’re not running a bureaucracy-we’re running a medical trial. And if you can’t tell the difference between a migraine and a stroke, you shouldn’t be near a clipboard.
It’s not about being strict. It’s about being smart. And right now? We’re being stupid.
Justin Fauth
February 11, 2026 AT 23:14Let’s be real-this whole system is a joke. We’re spending billions to report headaches while real problems slip through. And the FDA? They’re too busy chasing their own tail. Meanwhile, in China and India, they’re using AI to auto-classify events with 95% accuracy. We’re still having Zoom trainings with PowerPoint slides from 2015. This isn’t science-it’s bureaucracy with a lab coat.
And don’t tell me ‘when in doubt, report.’ That’s not safety-that’s cowardice. Report the right thing, or don’t report at all. I’m tired of playing Russian roulette with patient safety because someone didn’t want to get yelled at for under-reporting.
Daz Leonheart
February 13, 2026 AT 08:15One thing I’ve learned the hard way: if you’re unsure, report it. Then go back and ask why you were unsure. Was it the wording? The training? The protocol? Fix that. Because the next person shouldn’t have to guess. Safety isn’t about perfection-it’s about consistency. And consistency starts with you.
Mandy Vodak-Marotta
February 13, 2026 AT 18:59Okay, so I’ve been doing this for 12 years, and I’ve seen everything. I’ve seen people report a sneeze as an SAE because the patient said ‘it felt serious.’ I’ve seen people miss a cardiac arrhythmia because it was ‘just a flutter.’ And I’ve seen entire trials get shut down because of one misclassified event that turned out to be the tip of the iceberg.
Here’s what I tell my new team: forget the emotions. Forget the ‘it seemed bad.’ Just ask the four questions. Death? Life-threatening? Hospitalization? Disability? If the answer is yes to any of them? Report. If it’s no? Document. Don’t overthink it. Don’t over-report. Don’t under-report. Just follow the damn criteria.
And yes, AI helps. But I still read every report myself. Because no algorithm can understand the context of a 78-year-old with three stents who says, ‘I felt weird.’ That weird? That’s the signal. The algorithm won’t catch that. You have to.
Training isn’t optional. It’s the only thing keeping this from collapsing into chaos. And if your institution doesn’t have a clear policy? Make one. Write it down. Share it. Teach it. Because someone’s life depends on it-not on your gut feeling, not on your intuition, not on your ‘I’ve seen this before.’ On the rules. The clear, cold, hard rules.
And if you’re still confused? Call the sponsor. Call the IRB. Call me. I’ll answer. Because we’re all in this together. And no one should have to guess when it comes to safety.